
Surface energy

In surface science, surface energy (also interfacial free energy or surface free energy) quantifies the disruption of intermolecular bonds that occurs when a surface is created. In solid-state physics, surfaces must be intrinsically less energetically favorable than the bulk of the material (that is, the atoms on the surface must have more energy than the atoms in the bulk), otherwise there would be a driving force for surfaces to be created, removing the bulk of the material by sublimation. The surface energy may therefore be defined as the excess energy at the surface of a material compared to the bulk, or it is the work required to build an area of a particular surface. Another way to view the surface energy is to relate it to the work required to cut a bulk sample, creating two surfaces. There is "excess energy" as a result of the now-incomplete, unrealized bonding between the two created surfaces.
Cutting a solid body into pieces disrupts its bonds and increases the surface area, and therefore increases surface energy. If the cutting is done reversibly, then conservation of energy means that the energy consumed by the cutting process will be equal to the energy inherent in the two new surfaces created. The unit surface energy of a material would therefore be half of its energy of cohesion, all other things being equal; in practice, this is true only for a surface freshly prepared in vacuum. Surfaces often change their form away from the simple "cleaved bond" model just implied above. They are found to be highly dynamic regions, which readily rearrange or react, so that energy is often reduced by such processes as passivation or adsorption.
Assessment
[edit]Measurement
[edit]Contact angle
[edit]The most common way to measure surface energy is through contact angle experiments.[1] In this method, the contact angle of the surface is measured with several liquids, usually water and diiodomethane. Based on the contact angle results and knowing the surface tension of the liquids, the surface energy can be calculated. In practice, this analysis is done automatically by a contact angle meter.[2]
There are several different models for calculating the surface energy based on the contact angle readings.[3] The most commonly used method is OWRK, which requires the use of two probe liquids and gives out as a result the total surface energy as well as divides it into polar and dispersive components.
Contact angle method is the standard surface energy measurement method due to its simplicity, applicability to a wide range of surfaces and quickness. The measurement can be fully automated and is standardized.[4]
In general, as surface energy increases, the contact angle decreases because more of the liquid is being "grabbed" by the surface. Conversely, as surface energy decreases, the contact angle increases, because the surface doesn't want to interact with the liquid.
Other methods
[edit]The surface energy of a liquid may be measured by stretching a liquid membrane (which increases the surface area and hence the surface energy). In that case, in order to increase the surface area of a mass of liquid by an amount, δA, a quantity of work, γ δA, is needed (where γ is the surface energy density of the liquid). However, such a method cannot be used to measure the surface energy of a solid because stretching of a solid membrane induces elastic energy in the bulk in addition to increasing the surface energy.
The surface energy of a solid is usually measured at high temperatures. At such temperatures the solid creeps and even though the surface area changes, the volume remains approximately constant. If γ is the surface energy density of a cylindrical rod of radius r and length l at high temperature and a constant uniaxial tension P, then at equilibrium, the variation of the total Helmholtz free energy vanishes and we have

where F is the Helmholtz free energy and A is the surface area of the rod:

Also, since the volume (V) of the rod remains constant, the variation (δV) of the volume is zero, that is,

Therefore, the surface energy density can be expressed as

The surface energy density of the solid can be computed by measuring P, r, and l at equilibrium.
This method is valid only if the solid is isotropic, meaning the surface energy is the same for all crystallographic orientations. While this is only strictly true for amorphous solids (glass) and liquids, isotropy is a good approximation for many other materials. In particular, if the sample is polygranular (most metals) or made by powder sintering (most ceramics) this is a good approximation.
In the case of single-crystal materials, such as natural gemstones, anisotropy in the surface energy leads to faceting. The shape of the crystal (assuming equilibrium growth conditions) is related to the surface energy by the Wulff construction. The surface energy of the facets can thus be found to within a scaling constant by measuring the relative sizes of the facets.
Calculation
[edit]Deformed solid
[edit]In the deformation of solids, surface energy can be treated as the "energy required to create one unit of surface area", and is a function of the difference between the total energies of the system before and after the deformation:
- .

Calculation of surface energy from first principles (for example, density functional theory) is an alternative approach to measurement. Surface energy is estimated from the following variables: width of the d-band, the number of valence d-electrons, and the coordination number of atoms at the surface and in the bulk of the solid.[5][page needed]
Surface formation energy of a crystalline solid
[edit]In density functional theory, surface energy can be calculated from the following expression:

where
- Eslab is the total energy of surface slab obtained using density functional theory.
- N is the number of atoms in the surface slab.
- Ebulk is the bulk energy per atom.
- A is the surface area.
For a slab, we have two surfaces and they are of the same type, which is reflected by the number 2 in the denominator. To guarantee this, we need to create the slab carefully to make sure that the upper and lower surfaces are of the same type.
Strength of adhesive contacts is determined by the work of adhesion which is also called relative surface energy of two contacting bodies.[6][page needed] The relative surface energy can be determined by detaching of bodies of well defined shape made of one material from the substrate made from the second material.[7] For example, the relative surface energy of the interface "acrylic glass – gelatin" is equal to 0.03 N/m. Experimental setup for measuring relative surface energy and its function can be seen in the video.[8]
Estimation from the heat of sublimation
[edit]To estimate the surface energy of a pure, uniform material, an individual region of the material can be modeled as a cube. In order to move a cube from the bulk of a material to the surface, energy is required. This energy cost is incorporated into the surface energy of the material, which is quantified by:


where zσ and zβ are coordination numbers corresponding to the surface and the bulk regions of the material, and are equal to 5 and 6, respectively; a0 is the surface area of an individual molecule, and WAA is the pairwise intermolecular energy.
Surface area can be determined by squaring the cube root of the volume of the molecule:

Here, M̄ corresponds to the molar mass of the molecule, ρ corresponds to the density, and NA is the Avogadro constant.
In order to determine the pairwise intermolecular energy, all intermolecular forces in the material must be broken. This allows thorough investigation of the interactions that occur for single molecules. During sublimation of a substance, intermolecular forces between molecules are broken, resulting in a change in the material from solid to gas. For this reason, considering the enthalpy of sublimation can be useful in determining the pairwise intermolecular energy. Enthalpy of sublimation can be calculated by the following equation:

Using empirically tabulated values for enthalpy of sublimation, it is possible to determine the pairwise intermolecular energy. Incorporating this value into the surface energy equation allows for the surface energy to be estimated.
The following equation can be used as a reasonable estimate for surface energy:

Interfacial energy
[edit]The presence of an interface influences generally all thermodynamic parameters of a system. There are two models that are commonly used to demonstrate interfacial phenomena: the Gibbs ideal interface model and the Guggenheim model. In order to demonstrate the thermodynamics of an interfacial system using the Gibbs model, the system can be divided into three parts: two immiscible liquids with volumes Vα and Vβ and an infinitesimally thin boundary layer known as the Gibbs dividing plane (σ) separating these two volumes.

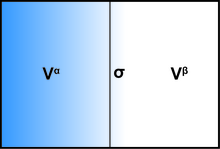
The total volume of the system is:

All extensive quantities of the system can be written as a sum of three components: bulk phase α, bulk phase β, and the interface σ. Some examples include internal energy U, the number of molecules of the ith substance ni, and the entropy S.

While these quantities can vary between each component, the sum within the system remains constant. At the interface, these values may deviate from those present within the bulk phases. The concentration of molecules present at the interface can be defined as:

where ciα and ciβ represent the concentration of substance i in bulk phase α and β, respectively.
It is beneficial to define a new term interfacial excess Γi which allows us to describe the number of molecules per unit area:

Wetting
[edit]Spreading parameter
[edit]Surface energy comes into play in wetting phenomena. To examine this, consider a drop of liquid on a solid substrate. If the surface energy of the substrate changes upon the addition of the drop, the substrate is said to be wetting. The spreading parameter can be used to mathematically determine this:

where S is the spreading parameter, γs the surface energy of the substrate, γl the surface energy of the liquid, and γs-l the interfacial energy between the substrate and the liquid.
If S < 0, the liquid partially wets the substrate. If S > 0, the liquid completely wets the substrate.[9]
Contact angle
[edit]A way to experimentally determine wetting is to look at the contact angle (θ), which is the angle connecting the solid–liquid interface and the liquid–gas interface (as in the figure).
- If θ = 0°, the liquid completely wets the substrate.
- If 0° < θ < 90°, high wetting occurs.
- If 90° < θ < 180°, low wetting occurs.
- If θ = 180°, the liquid does not wet the substrate at all.[10]
The Young equation relates the contact angle to interfacial energy:

where γs-g is the interfacial energy between the solid and gas phases, γs-l the interfacial energy between the substrate and the liquid, γl-g is the interfacial energy between the liquid and gas phases, and θ is the contact angle between the solid–liquid and the liquid–gas interface.[11]
Wetting of high- and low-energy substrates
[edit]The energy of the bulk component of a solid substrate is determined by the types of interactions that hold the substrate together. High-energy substrates are held together by bonds, while low-energy substrates are held together by forces. Covalent, ionic, and metallic bonds are much stronger than forces such as van der Waals and hydrogen bonding. High-energy substrates are more easily wetted than low-energy substrates.[12] In addition, more complete wetting will occur if the substrate has a much higher surface energy than the liquid.[13]
Modification techniques
[edit]The most commonly used surface modification protocols are plasma activation, wet chemical treatment, including grafting, and thin-film coating.[14][15][16] Surface energy mimicking is a technique that enables merging the device manufacturing and surface modifications, including patterning, into a single processing step using a single device material.[17]
Many techniques can be used to enhance wetting. Surface treatments, such as corona treatment,[18] plasma treatment and acid etching,[19] can be used to increase the surface energy of the substrate. Additives can also be added to the liquid to decrease its surface tension. This technique is employed often in paint formulations to ensure that they will be evenly spread on a surface.[20]
The Kelvin equation
[edit]As a result of the surface tension inherent to liquids, curved surfaces are formed in order to minimize the area. This phenomenon arises from the energetic cost of forming a surface. As such the Gibbs free energy of the system is minimized when the surface is curved.
The Kelvin equation is based on thermodynamic principles and is used to describe changes in vapor pressure caused by liquids with curved surfaces. The cause for this change in vapor pressure is the Laplace pressure. The vapor pressure of a drop is higher than that of a planar surface because the increased Laplace pressure causes the molecules to evaporate more easily. Conversely, in liquids surrounding a bubble, the pressure with respect to the inner part of the bubble is reduced, thus making it more difficult for molecules to evaporate. The Kelvin equation can be stated as:

where PK
0 is the vapor pressure of the curved surface, P0 is the vapor pressure of the flat surface, γ is the surface tension, Vm is the molar volume of the liquid, R is the universal gas constant, T is temperature (in kelvin), and R1 and R2 are the principal radii of curvature of the surface.
Surface modified pigments for coatings
[edit]Pigments offer great potential in modifying the application properties of a coating. Due to their fine particle size and inherently high surface energy, they often require a surface treatment in order to enhance their ease of dispersion in a liquid medium. A wide variety of surface treatments have been previously used, including the adsorption on the surface of a molecule in the presence of polar groups, monolayers of polymers, and layers of inorganic oxides on the surface of organic pigments.[21]
New surfaces are constantly being created as larger pigment particles get broken down into smaller subparticles. These newly-formed surfaces consequently contribute to larger surface energies, whereby the resulting particles often become cemented together into aggregates. Because particles dispersed in liquid media are in constant thermal or Brownian motion, they exhibit a strong affinity for other pigment particles nearby as they move through the medium and collide.[21] This natural attraction is largely attributed to the powerful short-range van der Waals forces, as an effect of their surface energies.
The chief purpose of pigment dispersion is to break down aggregates and form stable dispersions of optimally sized pigment particles. This process generally involves three distinct stages: wetting, deaggregation, and stabilization. A surface that is easy to wet is desirable when formulating a coating that requires good adhesion and appearance. This also minimizes the risks of surface tension related defects, such as crawling, cratering, and orange peel.[22] This is an essential requirement for pigment dispersions; for wetting to be effective, the surface tension of the pigment's vehicle must be lower than the surface free energy of the pigment.[21] This allows the vehicle to penetrate into the interstices of the pigment aggregates, thus ensuring complete wetting. Finally, the particles are subjected to a repulsive force in order to keep them separated from one another and lowers the likelihood of flocculation.
Dispersions may become stable through two different phenomena: charge repulsion and steric or entropic repulsion.[22] In charge repulsion, particles that possess the same like electrostatic charges repel each other. Alternatively, steric or entropic repulsion is a phenomenon used to describe the repelling effect when adsorbed layers of material (such as polymer molecules swollen with solvent) are present on the surface of the pigment particles in dispersion. Only certain portions (anchors) of the polymer molecules are adsorbed, with their corresponding loops and tails extending out into the solution. As the particles approach each other their adsorbed layers become crowded; this provides an effective steric barrier that prevents flocculation.[23] This crowding effect is accompanied by a decrease in entropy, whereby the number of conformations possible for the polymer molecules is reduced in the adsorbed layer. As a result, energy is increased and often gives rise to repulsive forces that aid in keeping the particles separated from each other.

Surface energies of common materials
[edit]| Material | Orientation | Surface energy (mJ/m2) |
|---|---|---|
| Polytetrafluoroethylene (PTFE) | 19[24][page needed] | |
| Glass | 83.4[25] | |
| Gypsum | 370[26] | |
| Copper | 1650[27] | |
| Magnesium oxide | (100) plane | 1200[28] |
| Calcium fluoride | (111) plane | 450[28] |
| Lithium fluoride | (100) plane | 340[28] |
| Calcium carbonate | (1010) plane | 230[28] |
| Sodium chloride | (100) plane | 300[29] |
| Sodium chloride | (110) plane | 400[30] |
| Potassium chloride | (100) plane | 110[29] |
| Barium fluoride | (111) plane | 280[28] |
| Silicon | (111) plane | 1240[28] |
See also
[edit]References
[edit]- ^ Marshall, S. J.; Bayne, S. C.; Baier, R.; Tomsia, A. P.; Marshall, G. W. (2010). "A review of adhesion science". Dental Materials. 26 (2): e11 – e16. doi:10.1016/j.dental.2009.11.157. PMID 20018362.
- ^ Laurén, S. "How To Measure Surface Free Energy?". blog.biolinscientific.com. Biolin Scientific. Retrieved 2019-12-31.
- ^ "Surface Free Energy: Measurements". biolinscientific.com. Biolin Scientific. Retrieved 2019-12-31.
- ^ "ISO 19403-2:2017. Paints and varnishes — Wettability — Part 2: Determination of the surface free energy of solid surfaces by measuring the contact angle". ISO. 2017.
- ^ Woodruff, D. P., ed. (2002). The Chemical Physics of Solid Surfaces. Vol. 10. Elsevier.[ISBN missing]
- ^ Contact Mechanics and Friction: Physical Principles and Applications. Springer. 2017. ISBN 9783662530801.
- ^ Popov, V. L.; Pohrt, R.; Li, Q. (September 2017). "Strength of adhesive contacts: Influence of contact geometry and material gradients". Friction. 5 (3): 308–325. doi:10.1007/s40544-017-0177-3.
- ^ Dept. of System Dynamics and Friction Physics (December 6, 2017). "Science friction: Adhesion of complex shapes". YouTube. Archived from the original on 2021-12-12. Retrieved 2018-01-28.
- ^ Bonn, D.; Eggers, J.; Indekeu, J.; Meunier, J.; Rolley, E. (2009). "Wetting and Spreading". Reviews of Modern Physics. 81 (2): 739–805. Bibcode:2009RvMP...81..739B. doi:10.1103/revmodphys.81.739.
- ^ Zisman, W. (1964). "Relation of the Equilibrium Contact Angle to Liquid and Solid Constitution". Contact Angle, Wettability, and Adhesion. Advances in Chemistry. Vol. 43. pp. 1–51. doi:10.1021/ba-1964-0043.ch001. ISBN 0-8412-0044-0.
- ^ Owens, D. K.; Wendt, R. C. (1969). "Estimation of the Surface Free Energy of Polymers". Journal of Applied Polymer Science. 13 (8): 1741–1747. doi:10.1002/app.1969.070130815.
- ^ De Gennes, P. G. (1985). "Wetting: statics and dynamics". Reviews of Modern Physics. 57 (3): 827–863. Bibcode:1985RvMP...57..827D. doi:10.1103/revmodphys.57.827.
- ^ Kern, K.; David, R.; Palmer, R. L.; Cosma, G. (1986). "Complete Wetting on 'Strong' Substrates: Xe/Pt(111)". Physical Review Letters. 56 (26): 2823–2826. Bibcode:1986PhRvL..56.2823K. doi:10.1103/physrevlett.56.2823. PMID 10033104.
- ^ Becker, H.; Gärtner, C. (2007). "Polymer microfabrication technologies for microfluidic systems". Analytical and Bioanalytical Chemistry. 390 (1): 89–111. doi:10.1007/s00216-007-1692-2. PMID 17989961. S2CID 13813183.
- ^ Mansky (1997). "Controlling Polymer-Surface Interactions with Random Copolymer Brushes". Science. 275 (5305): 1458–1460. doi:10.1126/science.275.5305.1458. S2CID 136525970.
- ^ Rastogi (2010). "Direct Patterning of Intrinsically Electron Beam Sensitive Polymer Brushes". ACS Nano. 4 (2): 771–780. doi:10.1021/nn901344u. PMID 20121228.
- ^ Pardon, G.; Haraldsson, T.; van der Wijngaart, W. (2016). "Surface Energy Mimicking: Simultaneous Replication of Hydrophilic and Superhydrophobic Micropatterns through Area-Selective Monomers Self-Assembly". Advanced Materials Interfaces. 3 (17): 1600404. doi:10.1002/admi.201600404. S2CID 138114323.
- ^ Sakata, I.; Morita, M.; Tsuruta, N.; Morita, K. (2003). "Activation of Wood Surface by Corona Treatment to Improve Adhesive Bonding". Journal of Applied Polymer Science. 49 (7): 1251–1258. doi:10.1002/app.1993.070490714.
- ^ Rosales, J. I.; Marshall, G. W.; Marshall, S. J.; Wantanabe, L. G.; Toledano, M.; Cabrerizo, M. A.; Osorio, R. (1999). "Acid-etching and Hydration Influence on Dentin Roughness and Wettability". Journal of Dental Research. 78 (9): 1554–1559. doi:10.1177/00220345990780091001. PMID 10512390. S2CID 5807073.
- ^ Khan, H.; Fell, J. T.; Macleod, G. S. (2001). "The influence of additives on the spreading coefficient and adhesion of a film coating formulation to a model tablet surface". International Journal of Pharmaceutics. 227 (1–2): 113–119. doi:10.1016/s0378-5173(01)00789-x. PMID 11564545.
- ^ a b c Wicks, Z. W. (2007). Organic Coatings: Science and Technology (3rd ed.). New York: Wiley Interscience. pp. 435–441.[ISBN missing]
- ^ a b Tracton, A. A. (2006). Coatings Materials and Surface Coatings (3rd ed.). Florida: Taylor and Francis Group. pp. 31-6 – 31-7.[ISBN missing]
- ^ Auschra, C.; Eckstein, E.; Muhlebach, A.; Zink, M.; Rime, F. (2002). "Design of new pigment dispersants by controlled radical polymerization". Progress in Organic Coatings. 45 (2–3): 83–93. doi:10.1016/s0300-9440(02)00048-6.
- ^ Kinloch, A. J. (1987). Adhesion & Adhesives: Science & Technology. London: Chapman & Hall.[ISBN missing]
- ^ Rhee, S.-K. (1977). "Surface energies of silicate glasses calculated from their wettability data". Journal of Materials Science. 12 (4): 823–824. Bibcode:1977JMatS..12..823R. doi:10.1007/BF00548176. S2CID 136812418.
- ^ Dundon, M. L.; Mack, E. (1923). "The Solubility and Surface Energy of Calcium Sulfate". Journal of the American Chemical Society. 45 (11): 2479–2485. doi:10.1021/ja01664a001.
- ^ Udin, H. (1951). "Grain Boundary Effect in Surface Tension Measurement". JOM. 3 (1): 63. Bibcode:1951JOM.....3a..63U. doi:10.1007/BF03398958.
- ^ a b c d e f Gilman, J. J. (1960). "Direct Measurements of the Surface Energies of Crystals". Journal of Applied Physics. 31 (12): 2208. Bibcode:1960JAP....31.2208G. doi:10.1063/1.1735524.
- ^ a b Butt, H.-J.; Graf, Kh.; Kappl, M. (2006). Physics and Chemistry of Interfaces. Weinheim: Wiley-VCH.[ISBN missing]
- ^ Lipsett, S. G.; Johnson, F. M. G.; Maass, O. (1927). "The Surface Energy and the Heat of Solution of Solid Sodium Chloride. I". Journal of the American Chemical Society. 49 (4): 925. doi:10.1021/ja01403a005.
External links
[edit]| Authority control databases: National |
|---|